Given a matrix of real RNA-seq counts, this function will
randomly assign samples to one of two groups, draw
the log2-fold change in expression between two groups for each gene,
and add this signal to the RNA-seq counts matrix. The user may specify
the proportion of samples in each group, the proportion of null genes
(where the log2-fold change is 0),
and the signal function. This is a specific application of the
general binomial thinning approach implemented in thin_diff.
Arguments
- mat
A numeric matrix of RNA-seq counts. The rows index the genes and the columns index the samples.
- prop_null
The proportion of genes that are null.
- signal_fun
A function that returns the signal. This should take as input
nfor the number of samples to return and then return only a vector of samples. Additional parameters may be passed throughsignal_params.- signal_params
A list of additional arguments to pass to
signal_fun.- group_prop
The proportion of individuals that are in group 1.
- corvec
A vector of target correlations.
corvec[i]is the target correlation of the latent group assignment vector with theith surrogate variable. The default is to set this toNULL, in which case group assignment is made independently of any unobserved confounding.- alpha
The scaling factor for the signal distribution from Stephens (2016). If \(x_1, x_2, ..., x_n\) are drawn from
signal_fun, then the signal is set to \(x_1 s_1^{\alpha}, x_2 s_2^{\alpha}, ..., x_n s_n^{\alpha}\), where \(s_g\) is the empirical standard deviation of gene \(g\). Setting this to0means that the effects are exchangeable, setting this to1corresponds to the p-value prior of Wakefield (2009). You would rarely set this to anything but0or1.- permute_method
Should we use the Gale-Shapley algorithm for stable marriages (
"marriage") (Gale and Shapley, 1962) as implemented in the matchingR package, or the Hungarian algorithm (Papadimitriou and Steiglitz, 1982) ("hungarian") as implemented in the clue package (Hornik, 2005)? The Hungarian method almost always works better, so is the default.- type
Should we apply binomial thinning (
type = "thin") or just naive multiplication of the counts (type = "mult"). You should always have this set to"thin".
Value
A list-like S3 object of class ThinData.
Components include some or all of the following:
matThe modified matrix of counts.
designmatThe design matrix of variables used to simulate signal. This is made by column-binding
design_fixedand the permuted version ofdesign_perm.coefmatA matrix of coefficients corresponding to
designmat.design_obsAdditional variables that should be included in your design matrix in downstream fittings. This is made by column-binding the vector of 1's with
design_obs.svA matrix of estimated surrogate variables. In simulation studies you would probably leave this out and estimate your own surrogate variables.
cormatA matrix of target correlations between the surrogate variables and the permuted variables in the design matrix. This might be different from the
target_coryou input because we pass it throughfix_corto ensure positive semi-definiteness of the resulting covariance matrix.matching_varA matrix of simulated variables used to permute
design_permif thetarget_coris notNULL.
Details
The specific application of binomial thinning to the two-group model was used in Gerard and Stephens (2018) and Gerard and Stephens (2021). This is a specific case of the general method described in Gerard (2020).
References
Gale, David, and Lloyd S. Shapley. "College admissions and the stability of marriage." The American Mathematical Monthly 69, no. 1 (1962): 9-15. doi: 10.1080/00029890.1962.11989827 .
Gerard, D., and Stephens, M. (2021). "Unifying and Generalizing Methods for Removing Unwanted Variation Based on Negative Controls." Statistica Sinica, 31(3), 1145-1166 doi: 10.5705/ss.202018.0345 .
David Gerard and Matthew Stephens (2018). "Empirical Bayes shrinkage and false discovery rate estimation, allowing for unwanted variation." Biostatistics, doi: 10.1093/biostatistics/kxy029 .
Gerard, D (2020). "Data-based RNA-seq simulations by binomial thinning." BMC Bioinformatics. 21(1), 206. doi: 10.1186/s12859-020-3450-9 .
Hornik K (2005). "A CLUE for CLUster Ensembles." Journal of Statistical Software, 14(12). doi: 10.18637/jss.v014.i12 . doi: 10.18637/jss.v014.i12 .
C. Papadimitriou and K. Steiglitz (1982), Combinatorial Optimization: Algorithms and Complexity. Englewood Cliffs: Prentice Hall.
Stephens, Matthew. "False discovery rates: a new deal." Biostatistics 18, no. 2 (2016): 275-294. doi: 10.1093/biostatistics/kxw041 .
Wakefield, Jon. "Bayes factors for genome-wide association studies: comparison with P-values." Genetic epidemiology 33, no. 1 (2009): 79-86. doi: 10.1002/gepi.20359 .
See also
select_countsFor subsampling the rows and columns of your real RNA-seq count matrix prior to applying binomial thinning.
thin_diffFor the more general thinning approach.
ThinDataToSummarizedExperimentFor converting a ThinData object to a SummarizedExperiment object.
ThinDataToDESeqDataSetFor converting a ThinData object to a DESeqDataSet object.
Examples
## Simulate data from given matrix of counts
## In practice, you would obtain Y from a real dataset, not simulate it.
set.seed(1)
nsamp <- 10
ngene <- 1000
Y <- matrix(stats::rpois(nsamp * ngene, lambda = 50), nrow = ngene)
thinout <- thin_2group(mat = Y,
prop_null = 0.9,
signal_fun = stats::rexp,
signal_params = list(rate = 0.5))
## 90 percent of genes are null
mean(abs(thinout$coef) < 10^-6)
#> [1] 0.9
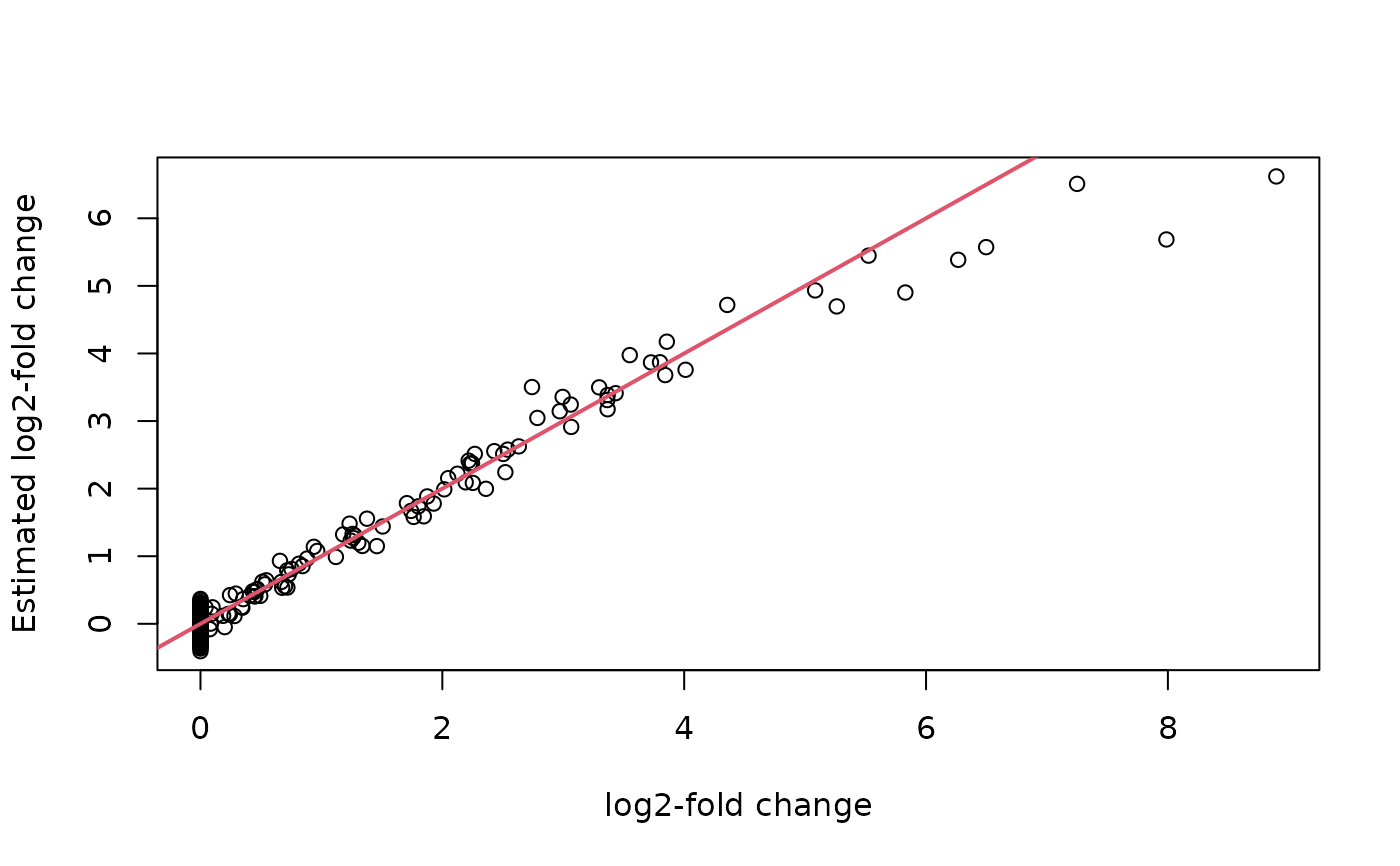
## Check the estimates of the log2-fold change
Ynew <- log2(t(thinout$mat + 0.5))
X <- thinout$designmat
Bhat <- coef(lm(Ynew ~ X))["X", ]
plot(thinout$coefmat,
Bhat,
xlab = "log2-fold change",
ylab = "Estimated log2-fold change")
abline(0, 1, col = 2, lwd = 2)